
L’apprentissage profond replie les protéines « in silico »
DeepMind devance largement ses compétiteurs dans la course au repliement numérique des protéines. L’apprentissage profond est désormais la voie royale pour prédire la structure tridimensionnelle des protéines et leurs interactions.
La biologie « in silico » a fait un grand pas en avant en décembre dernier. La technologie AlphaFold2 de la société DeepMind (filiale d’Alphabet et Google), faisant appel au dernier cri de l’apprentissage profond, a remporté haut la main une compétition internationale où s’affrontaient une centaine d’équipes internationales d’un sport méconnu : le repliement de protéine. Ou plutôt, la prédiction numérique de la structure tridimensionnelle de protéines à partir de leur séquence linéaire. Ce qui devrait avoir un impact notable sur les progrès à venir en médecine et biologie.
Le ribosome, une sorte d’imprimante moléculaire
Les protéines sont des macromolécules constituées d’une chaîne de plus petites molécules : les acides aminés. Sauf exception, chaque chaînon est issu d’un catalogue de vingt acides aminés « standards ». La liste des briques constituant ainsi une protéine est codée par son gène, un fragment du patrimoine génétique, c’est-à-dire de l’ADN de l’espèce considérée. L’histoire d’une protéine commence lorsque ce gène est transcrit en un petit brin d’ARN messager (ARNm). Cette copie temporaire est ensuite prise en charge par un ribosome, une sorte d’imprimante moléculaire capable de lire le code génétique porté par un ARN messager et de synthétiser la séquence d’acides aminés correspondante.
Mais après sa sortie de l’usine ribosome, une protéine ne reste pas longtemps sous la forme d’une chaîne linéaire : à peine fabriquée, elle se replie sur elle-même sous l’effet d’interactions diverses entre ses constituants, et forme une sorte de pelote. À l’heure où la lecture intégrale du génome d’un individu est devenue monnaie courante, obtenir la séquence linaire d’une protéine est un jeu d’enfant. En revanche, déterminer expérimentalement la structure tridimensionnelle de cette même protéine reste un processus laborieux, faisant appel à des techniques coûteuses, comme la cristallographie aux rayons X, la spectroscopie RMN et la cryo-microscopie électronique.
Calculer la conformation 3D des molécules
Or, c’est précisément la structure tridimensionnelle d’une protéine, sa conformation, qui détermine ses capacités d’interaction avec d’autres molécules, et donc sa fonction. Voilà pourquoi, dès les années 1970, on a tenté de déterminer la conformation en 3D des protéines par le calcul. Depuis 1994, les progrès de ce nouveau sport se mesurent au cours d’une compétition internationale bisannuelle : CASP, pour « Critical Assessment of protein Structure Prediction » (Évaluation critique de la prédiction de la structure des protéines). Règle du jeu : une liste de protéines dont la structure 3D vient d’être déterminée expérimentalement, mais n’a pas encore été publiée, est proposée aux participants. Lesquels font alors travailler leurs algorithmes à partir de la seule séquence linéaire de ces protéines et rendent à l’heure dite leur copie : des prédictions de structure 3D. La comparaison avec les résultats expérimentaux permet alors d’évaluer les performances de chaque équipe et d’établir un palmarès.
Déjà en 2018, DeepMind avait emporté la palme d’or de cette épreuve avec son premier AlphaFold. Mais en décembre 2020, une version nettement améliorée, AlphaFold2, a littéralement surclassé tous ses concurrents. « Ces résultats constituent sans aucun doute un exploit historique », estime Sergei Grudinin. Ce chercheur CNRS de l’équipe Nano-D (CNRS, Inria, université de Grenoble) de Minatec a joué un rôle pionnier dans le recours aux réseaux de neurones en biologie structurale.
Évaluer les performances des prédictions de structures
Plusieurs métriques sont utilisées pour évaluer les performances des prédictions de structures tridimensionnelles de protéines faites par les équipes concurrentes. La plus commune s’appelle GDT_TS, pour « Global Distance Test, Total Score » (soit : test de distance global, score total). En simplifiant, elle mesure la proportion d’acides aminés « bien » placés par la prédiction, par rapport au modèle tridimensionnel déterminé par des méthodes expérimentales. « AlphaFold2 a dépassé la barre des 90% en moyenne selon ce critère, ce qui est une performance remarquable », précise le chercheur. Avec un score médian de 92,4% le poulain de DeepMind n’a été devancé par un concurrent que 9 fois sur 97 protéines.
On se représente mieux l’exploit en faisant appel à une autre métrique, qui mesure la distance moyenne des atomes du modèle prédit par rapport à leur position déterminée expérimentalement : le RMSD (Root Mean Square Deviation), c’est à dire l’écart quadratique moyen, sous-entendu entre les positions atomiques. On considère souvent les atomes « carbone alpha », autour desquels s’articulent les chaînes d’acides aminés. « AlphaFold2 obtient une précision de 1,5 angström une fois sur deux, indique Sergei Grudinin. » Or, 1,5 angström (soit 0,15 nanomètre), c’est à peu près la distance moyenne entre deux carbones du squelette de ces molécules… Et même en considérant tous les atomes, AlphaFold2 détermine leur position avec un RMSD inférieur à 3 angströms trois fois sur quatre.
Algorithmes de dynamique moléculaire
« Les premiers efforts pour déterminer par le calcul la conformation 3D d’une protéine remontent à la fin des années 1970, explique Élodie Laine, enseignante-chercheuse au sein de l’équipe Analytical Genomics (Génomique analytique) du Laboratory of Computational and Quantitative Biology (Laboratoire de biologie computationnelle et quantitative, LCQB, Sorbonne Université, CNRS). À l’époque, ces travaux relevaient de la simulation numérique. On développait des algorithmes de dynamique moléculaire, qui modélisaient les mouvements des atomes et l’effet des diverses forces en jeu. » Cette approche nécessitait d’énormes puissances de calcul et l’on faisait donc appel à des supercalculateurs. La puissance de ces derniers ne cessant de croître, on a commencé à obtenir des résultats intéressants dans les années 1990.
Consécration du Deep Learning
« Dans les années 2000, on conçoit des architectures matérielles spécialisées, poursuit Élodie Laine, et on recode les logiciels pour exploiter les cartes graphiques, désormais plus accessibles. Et les algorithmes connaissent d’importantes innovations. C’est dans les années 2010 que l’on commence à faire appel à l’apprentissage profond. Les premiers réseaux de neurones profonds font leur apparition à CASP10, l’édition 2012 de la compétition CASP. Ils connaissent un succès croissant à CASP11 (cru 2014) et CASP12 (cru 2016). Et en 2018 (CASP13), c’est la consécration du deep learning. » DeepMind emporte alors la compétition avec son premier AlphaFold.
« Le repliement de protéines n’est pas le seul problème important en biologie structurale auquel l’apprentissage profond est désormais en mesure d’apporter des réponses, précise Sergei Grudinin. Déterminer la façon dont les protéines, et d’autres molécules, s’arriment l’une à l’autre, ce que l’on appelle le « docking », est également un sujet très important où des progrès notables sont enregistrés. » Pouvoir découvrir par le seul calcul, et donc potentiellement massivement, non seulement la conformation des protéines mais aussi la façon dont elles interagissent, ouvre de vastes perspectives pour la biologie et la médecine. On pense immédiatement à la découverte de nouveaux médicaments, mais aussi à la conception de protéines nouvelles pour les secteurs de l’agriculture, de l’agroalimentaire, voire de l’énergie.
Pierre Vandeginste
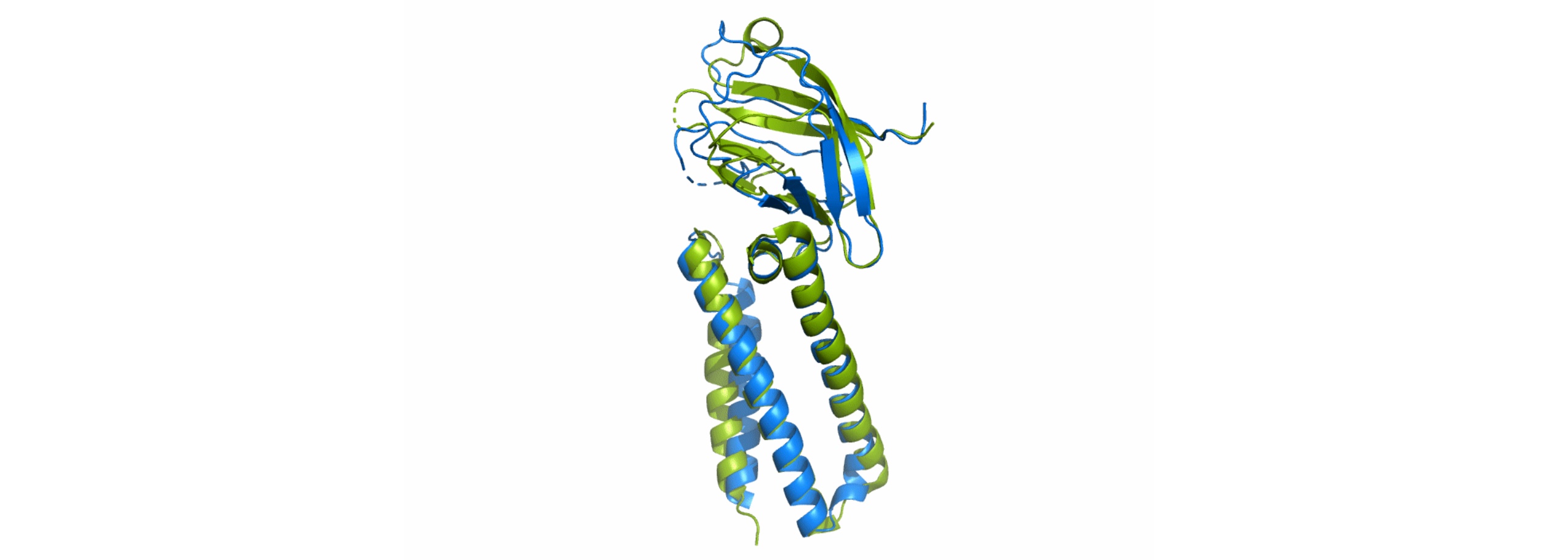
Image de Une : La protéine ORF3a du virus SARS-CoV-2. En bleu : le modèle prédit par AlphaFold. En vert : la structure déterminée expérimentalement. https://deepmind.com/research/open-source/computational-predictions-of-protein-structures-associated-with-COVID-19 © DeepMind


