La lutte contre le virus SARS-CoV-2 commence in silico
⏱ 6 minLes sciences des données sont désormais tout naturellement à l’avant-garde de la lutte contre le Covid-19. Elles permettent de modéliser la pandémie, de proposer des outils de dépistage et de diagnostic, mais encore et surtout d’accélérer la recherche de thérapies.
La loi de Moore, alliée au parallélisme massif, a permis de concevoir des supercalculateurs offrant des puissances qui se mesurent désormais en pétaflops (millions de milliards d’opérations flottantes par seconde). De telles puissances de calcul autorisent la modélisation en 3D de la structure de molécules complexes, d’assemblages de molécules, voire d’un virus entier. Mieux, on a développé des algorithmes capables de simuler les interactions entre un virus et des molécules susceptibles de s’y fixer (on parle de « docking »), et ainsi d’inhiber un processus (pénétration dans la cellule hôte, transcription, réplication…) jouant un rôle essentiel dans le mécanisme d’infection.
Ces simulations permettent de sélectionner « in silico » des molécules susceptibles de déboucher sur une approche phamacologique. On parle de criblage virtuel (« virtual screening », en anglais). Il n’est pas question, pour l’heure, de « découvrir » directement, par le seul calcul, le médicament qui nous tirera d’affaire, mais de faire le tri, de sélectionner dans des bases de données de molécules une liste de candidats dignes d’intérêt. Ensuite, bien sûr, le chemin de croix de la pharmacologie suit son cours. Les candidats repérés in silico seront passés au crible des tests in vitro, puis des tests in vivo, sur l’animal, pour finalement passer devant le juge de paix des essais cliniques, sur l’homme.
De nombreuses initiatives ont été lancées ces derniers mois pour utiliser ces nouvelles méthodes de simulation pour lutter contre le SARS-CoV-2, responsable de la pandémie actuelle. Ainsi l’initiative HT-Covid, pilotée par une équipe de l’université de Reims, consiste en un criblage à très grande échelle de molécules simples : d’une part un catalogue de 70 000 composés existants, d’autre part un milliard et demi de molécules « facilement synthétisables », proposées par un algorithme relevant de l’intelligence artificielle.
Simuler la dynamique des interactions moléculaires
Un outil de simulation ne produit qu’une approximation de la réalité. Le degré de réalisme dépend de l’échelle à laquelle il travaille et de sa maîtrise plus ou moins exhaustive des phénomènes physiques en jeu. Simuler statiquement des interactions moléculaires, c’est déjà un petit exploit, qui peut donner des résultats intéressants, toutefois on peut aller encore plus loin, pour mieux toucher du doigt la réalité biologique.
L’initiative Covid-HP, portée par Sorbonne Université et impliquant quatre équipes de recherche françaises et trois internationales, vise à réaliser un criblage virtuel avec une précision inégalée, pour accélérer la découverte de traitements contre le Covid-19. Cela grâce à une modélisation dynamique et tenant compte de certains effets quantiques des interactions entre virus et molécules potentiellement thérapeutiques. C’est le premier projet lié au Covid-19 accepté dans le cadre de la procédure d’urgence mise en place par le Partenariat pour le calcul avancé en Europe (Prace). Covid-HP est également soutenu par le Genci (Grand équipement national de calcul intensif), qui lui accorde un accès privilégié au supercalculateur Jean Zay, de l’Idris (Institut du développement et des ressources en informatique scientifique).
Covid-HP est piloté par le directeur du Laboratoire de chimie théorique de Sorbonne Université, Jean-Philip Piquemal. Ce projet collaboratif implique également les équipes françaises de Matthieu Montes (CNAM), Maxime Maria (XLIM, université de Limoges) et Michael Nilges (Structural Bioinformatics, Institut Pasteur), ainsi que trois équipes états-uniennes : Pengyu Ren (Dept of Biomedical Engineering, University of Texas at Austin), Jay Ponder (Dept of Chemistry, Washington University in Saint Louis, Missouri) et Andrés Cisneros (Dept of Chemistry, University of North Texas).
« Pour approcher de plus près la réalité, explique Jean-Philip Piquemal, il faut tout d’abord tenir compte du fait qu’une grosse molécule se courbe, se tord en permanence, et qu’a fortiori un assemblage de molécules complexes comme un virus ne cesse de se déformer. De ce fait, une simulation statique de l’interaction entre une molécule éventuellement thérapeutique et une région d’un virus peut être instructive, mais reste forcément approximative. Pour approcher un peu plus la réalité chimique de l’interaction, il faut tout d’abord introduire la dimension temporelle, simuler dynamiquement ces molécules. »
Tenir compte d’effets relevant de la physique quantique
Il s’agit en quelque sorte de passer de la photo de la rencontre à la vidéo. « Mais nous faisons encore mieux, ajoute Jean-Philip Piquemal, car l’outil que nous avons créé, et que nous améliorons depuis dix ans, Tinker-HP, ne se contente pas de simuler de manière classique (c’est-à-dire en utilisant une physique newtonienne à deux corps) les forces régissant les interactions moléculaires, comme les liaisons de covalence ou les forces de van der Waals simples. Lorsque deux molécules sont très proches, des interactions plus complexes, typiques de la physique quantique (comme les « effets à N corps »), interviennent. Tinker-HP nous permet non seulement de modéliser ces interactions moléculaires dynamiquement à l’échelle de l’atome, mais également de tenir compte de certains effets quantiques. Covid-HP est la seule initiative à travailler à cette échelle. Cela doit nous permettre d’obtenir des informations plus fiables, plus prédictives sur ces interactions. »
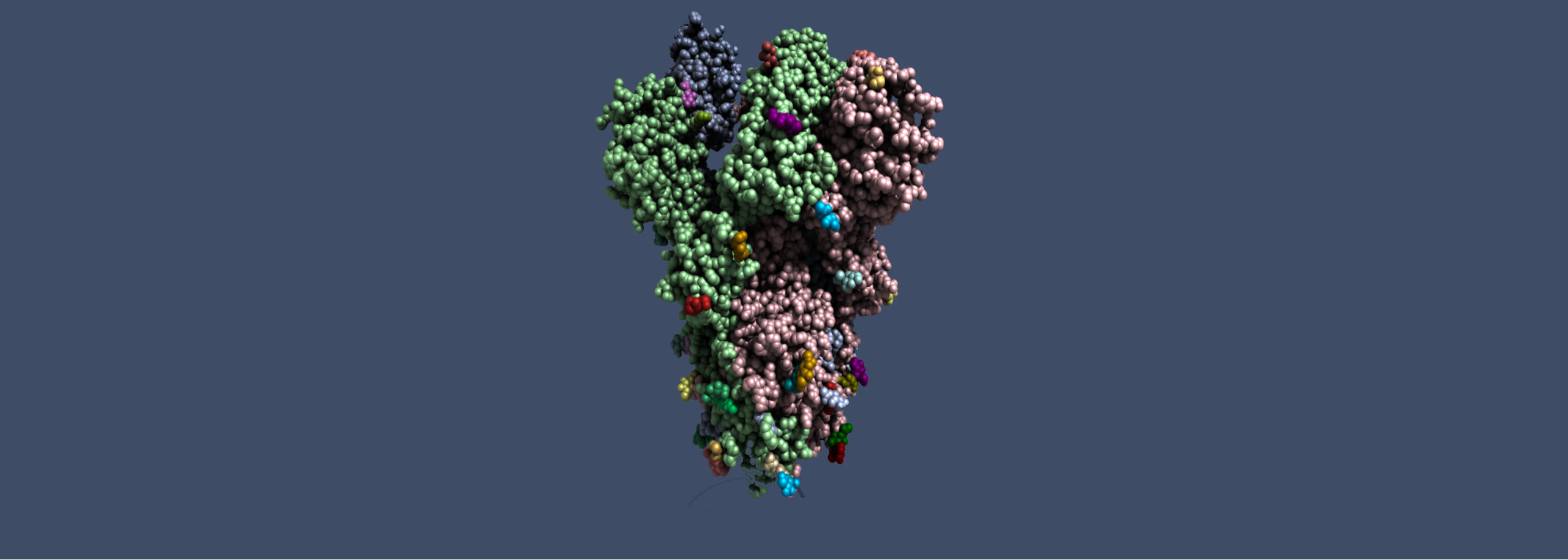
Avant la pandémie, la cible essentielle de son équipe était le VIH. « Nous avons donc une certaine expérience des virus. Nous avons introduit dans notre système les modèles les plus précis du SARS-CoV-2 et de ses composants au fur et à mesure qu’ils étaient publiés. Le virus comprend 250 millions d’atomes, les régions intéressantes représentent entre un demi et trois millions d’atomes. Nous disposons notamment d’un modèle à très haute résolution du fameux « spike » (spicule), dont le virus possède un grand nombre d’exemplaires à sa surface, et qui lui permet de s’arrimer au récepteur ACE2 des cellules épithéliales des voies respiratoires. C’est notre première cible. Et dans un premier temps, nous chercherons des molécules susceptibles d’interférer avec ce mécanisme dans des bases de données de médicaments déjà sur le marché. Car le repositionnement de médicaments déjà bien connus (drug repurposing) est le moyen le plus efficace de trouver une arme utilisable rapidement. »
Au CNAM, Matthieu Montes, responsable de l’équipe Molecular modeling and drug design, est impliqué notamment dans l’aspect visualisation du projet. « Pour simuler au plus près une interaction entre une molécule thérapeutique potentielle et une région du virus, il faut en quelque sorte filmer cette rencontre virtuelle, calculer une image par femtoseconde, pendant quelques picosecondes, nanosecondes ou microsecondes. Sachant qu’entre deux images, le virus se déforme, la molécule se déplace, tourne sur elle-même et qu’entre les deux circulent des dizaines de milliers de molécules d’eau… On simule un système composé typiquement d’un million d’atomes. Et alors qu’en modélisation moléculaire classique, les atomes sont de simples boules et les liaisons atomiques des ressorts, dans nos modèles chaque atome prend en compte le fait qu’en physique quantique les noyaux et électrons interfèrent avec leurs voisins. Les modèles de champs de forces polarisables que nous utilisons ne résolvent certes pas une équation de Schrödinger mais ils sont capables de la mimer efficacement et demeurent très rapides grâce aux supercalculateurs. Ils offrent ainsi la possibilité de simuler à large échelle de grands systèmes biologiques et donnent accès à une prédiction plus fidèle des interactions moléculaires. »
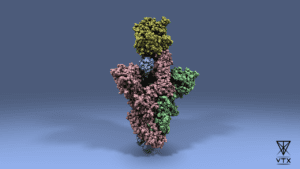
Spike du SARS-CoV-2 lié au récepteur ACE2 (en jaune), présent sur la membrane des cellules épithéliales des voies respiratoires, image réalisée avec VTX, M. Montes (Cnam), M. Maria (Univ. Limoges).
Cartographier toutes les interactions impliquées dans le Covid-19
Une autre initiative, baptisée Covid-19 Disease Map, ne se focalise pas sur le virus, mais étudie le problème Covid-19 avec un objectif grand angle, ou plutôt avec un zoom, puisqu’elle s’intéresse à la pathologie à toutes les échelles, depuis les plus petites molécules impliquées jusqu’à l’organisme entier. Son approche relève de la « biologie des systèmes » (mais on parle parfois de « biologie des réseaux »). Cette offensive internationale mobilise 150 chercheurs dans 24 pays : France, Luxembourg, Allemagne, Espagne, Pays-Bas, Royaume-Uni, Italie… mais aussi Japon, USA, Canada, Australie, et Afrique du sud. Elle a publié le 5 mai dans Nature Scientific Data un véritable plan de bataille.
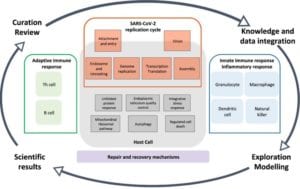
D’après Ostaszewski et al. COVID-19 Disease Map, building a computational repository of SARS-CoV-2 virus-host interaction mechanisms. Sci Data 7, 136 (2020). doi.org/10.1038/s41597-020-0477-8
À l’Institut Curie, à Paris, Emmanuel Barillot dirige l’unité Cancer et génome : bio-informatique, biostatistiques et épidémiologie, qui applique d’habitude ce type d’approche essentiellement en cancérologie. « L’idée est de cartographier toutes les interactions, les voies de signalisation, les échanges impliqués dans la pathologie, explique-t-il. Les modèles utilisés font intervenir toutes sortes d’acteurs : molécules petites et grandes (protéines, anticorps, gènes, enzymes, hormones…), cellules, tissus, organes. Ils sont construits à partir de l’analyse de la littérature scientifique par des experts de toutes disciplines, notamment à l’aide d’outils de fouille de données. »
« Nous disposons alors d’un modèle conceptuel de la pathologie, ajoute Inna Kuperstein, une chercheuse de l’Institut Curie très impliquée dans ce projet, une représentation des mécanismes en jeu. Une description, à l’aide de formalismes standardisés, de tout ce que l’on sait sur les voies interconnectées de signalisation, métaboliques et de régulation des gènes. L’objectif est ensuite de faire « tourner » ces modèles à l’aide d’outils mathématiques permettant d’y repérer des chemins critiques, des points où une intervention pharmacologique aurait de bonnes chances d’être efficace. »
« Dans un premier temps, précise Cristobal Monraz, un jeune chercheur également impliqué dans le projet, nous mettons l’accent sur le cycle de réplication du virus et la transcription. Mais nous avons également commencé à travailler sur d’autres aspects de la pathologie, comme son impact sur la pression artérielle pulmonaire. » Des milliers de chercheurs, à travers le monde, étudient cette maladie émergente sous toutes ses coutures. On dénombrait à la mi-mai quelque treize mille publications sur le sujet. Et le flux ne tarit pas. Les connaissances ne cessent de s’accumuler pour compléter cette carte du Covid-19 et en faire un jour un outil efficace pour le juguler.
Image à la une : Spike du SARS-CoV-2, image réalisée avec VTX, M. Montes (Cnam), M. Maria (Univ. Limoges).